General workflow
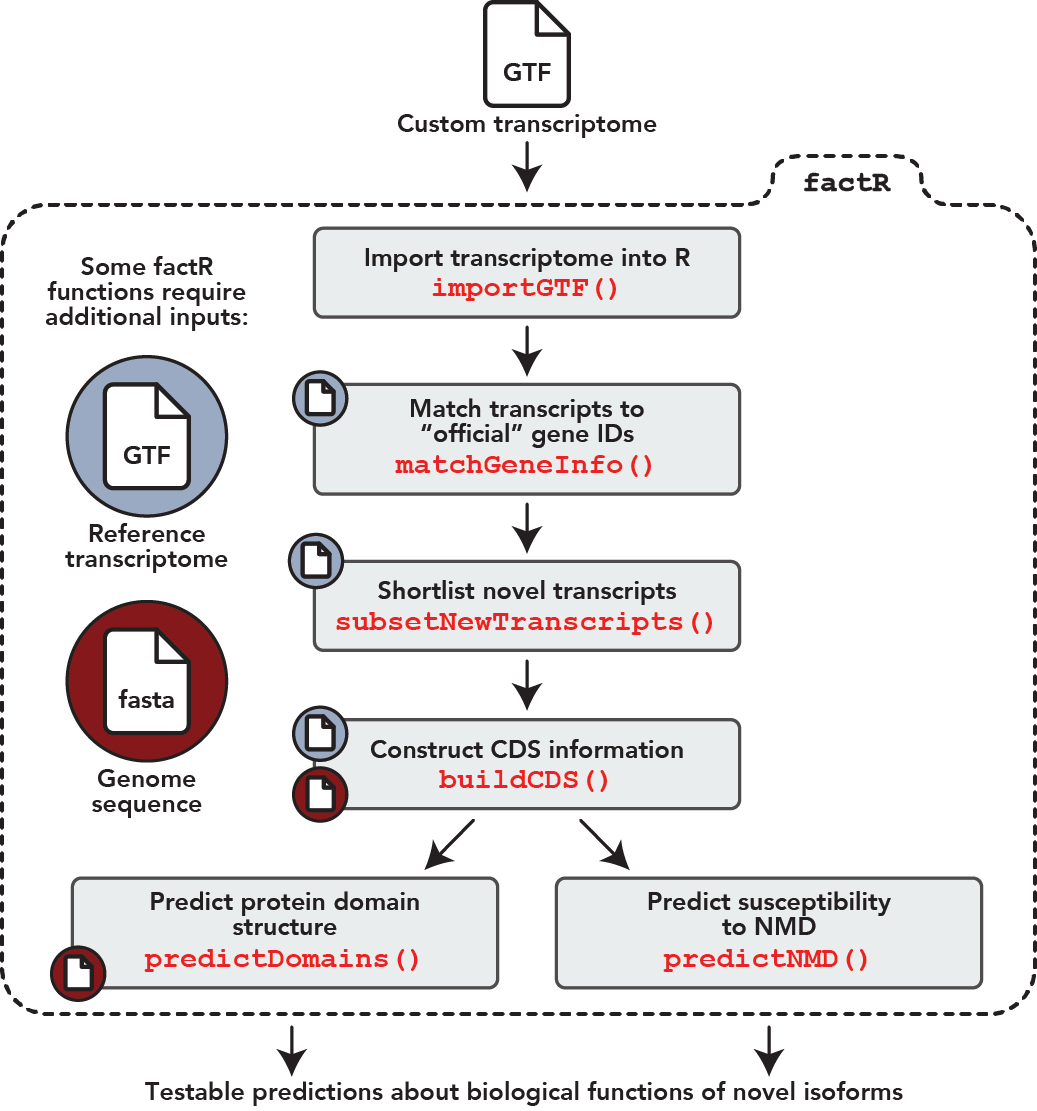
factR is a robust and easy-to-use R package with tools to process custom-assembled transcriptomes (GTF). Below are factR’s key functions:
- Core features
- Construct transcript coding (CDS) information using a reference-guided process
- Predict protein domains on coding transcripts
- Predict sensitivity of coding transcripts to Nonsense-mediated decay
- Supporting features
- Match chromosome levels of query GTF/object to reference annotation
- Match gene_id and gene_names of query GTF to reference annotation
- Plot transcripts from GTF GRanges object using wiggleplotr
- Subset new transcripts from custom transcriptome
How to install
The latest stable version can be installed directly from Bioconductor:
if (!requireNamespace("BiocManager", quietly=TRUE))
install.packages("BiocManager")
BiocManager::install("factR")Alternatively, you may install the development version of factR using devtools:
# install.packages("devtools")
devtools::install_github("fursham-h/factR")Getting started
See our quickstart guide or our full vignette on how to get started
Acknowledgements
We thank Kaur Alasoo for sharing code resources for wiggleplotr and for valuable discussions on the design of the package.
Citing factR
Please cite the following references if you use factR:
- Fursham Hamid, Kaur Alasoo, Jaak Vilo, Eugene Makeyev (2022); Functional annotation of custom transcriptomes; Methods in Molecular Biology
- Fursham Hamid (2022); Functional Annotation of Custom Transcriptomes; Bioconductor